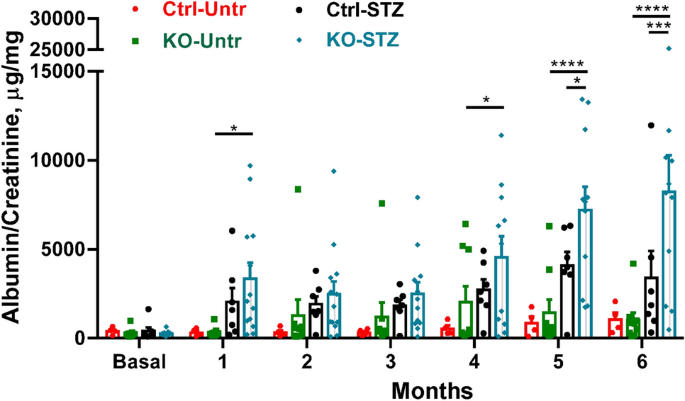
The present study shows that deletion of IRE1α in podocytes exacerbates albuminuria in experimental type I diabetic nephropathy. While albuminuria increased in diabetic control mice, albuminuria was significantly exaggerated in diabetic IRE1α KO mice over a 6-month period. Furthermore, after 6 months of diabetes, the number of podocytes per glomerulus and the expression of synaptopodin was comparable among untreated (non-diabetic) control, untreated IRE1α KO and diabetic control mice, but diabetic IRE1α KO mice showed a significant reduction. Deletion of IRE1α in podocytes exacerbated ultrastructural glomerular injury in diabetic nephropathy. Foot processes and the GBM demonstrated widening in diabetic IRE1α KO mice, but not in diabetic controls. Disruption of the podocyte plasma membranes, significantly dilated ER and prominent mitochondrial damage were evident only in diabetic IRE1α KO mice, although the changes were not widespread. Therefore, in diabetic nephropathy, IRE1α is important in preserving podocyte structure and function, and interestingly, IRE1α preserves not only ER integrity, but also mitochondrial structure. Although diabetic IRE1α KO mice showed widened GBMs, we did not detect changes in glomerular collagen IV expression. Possibly these mice had increases in other collagen isoforms, such as collagen I (interstitial collagen).
Since IRE1α is a transducer of the UPR and was shown to be linked to autophagy5, we investigated if these processes were affected in diabetic nephropathy. The ER chaperones GRP94 and MANF, as well as the marker of autophagy LC3-II (and total LC3 levels), were increased in glomeruli of diabetic control mice, but not diabetic IRE1α KO mice. Conversely, the autophagy substrate p62 was increased in diabetic IRE1α KO mice, but not in the diabetic control group. Thus, IRE1α mediated activation of the UPR and autophagy, as well as production of LC3 in diabetes. The apparent increased podocyte injury in IRE1α KO mice is at least in part attributable to impaired UPR and autophagy. Since diabetic IRE1α KO mice showed mitochondrial damage, we examined glomerular expression PGC1α; however, we did not observe consistent changes. Thus, loss of IRE1α did not apparently affect mitochondrial biogenesis, although impaired mitochondrial biogenesis has been observed in diabetic kidneys35. The presence of damaged mitochondria, in view of the impaired autophagy, may reflect a disruption of their clearance by mitophagy12.
The results of our study, which demonstrates an adaptive/protective role of IRE1α in type I diabetic nephropathy are in keeping with an earlier report40. In this earlier study, mice with STZ-induced diabetes were followed for 12 weeks, and deletion of IRE1α in podocytes worsened albuminuria at 2–12 weeks. At 12 weeks, diabetic IRE1α KO mice also showed wider foot processes and GBM, podocyte loss and cleavage of caspase-3 compared with diabetic control mice. The study focused on the expression of alcohol dehydrogenase-1, which was reduced in both untreated and diabetic IRE1α KO mice compared with respective control groups. The authors concluded that the mechanisms of interaction between IRE1α and alcohol dehydrogenase-1 will require further investigation. Parameters of ER stress, the UPR or autophagy were not examined, nor was deletion of IRE1α in podocytes.
A number of other studies have addressed parameters of ER stress in experimental type 1 or 2 diabetes. The results have been variable and these have been reviewed elsewhere4,22,25. Hyperglycemia, free fatty acids and advanced glycation end products can induce ER chaperones, sXBP1, C/EBP homologous protein (CHOP; a pro-apoptotic transcription factor) and oxidative stress, and can induce apoptosis in renal cells22,25. Outcomes appear to depend on renal cell type and context. In podocytes, diabetic metabolic changes were shown to perturb the balance between the UPR, autophagy and mTOR signaling22,41,42.
In STZ diabetes, increased levels of the ER chaperone BiP, phospho-PERK (reflecting PERK activation), CHOP and caspase-12 were reported in glomerular and tubular cells, along with enhanced apoptosis43. Moreover, in this model, CHOP KO mice showed less proteinuria, compared with CHOP-replete controls44. Recently, combined intervention with CHOP-antisense oligonucleotides and angiotensin-converting enzyme inhibition reduced glomerular and tubular damage in type 2 diabetic nephropathy in db/db mice45, in keeping with the result in CHOP KO mice44. In db/db mice, ER stress triggered the expression of inflammatory genes46. Mice with diabetic nephropathy (either induced by STZ or in db/db mice) showed increased levels of CHOP and activated ATF6 in the renal cortex. In these mice, translocation of sXBP1 into the nucleus was impaired24. Lowering of blood glucose or treatment with a chemical chaperone (to improve ER protein folding) normalized sXBP1, ATF6 and CHOP levels, and reduced albuminuria and renal damage associated with diabetes. The severity of diabetic nephropathy and levels of CHOP and ATF6 were exaggerated in podocyte-specific Xbp1-knockout mice and in transgenic mice overexpressing ATF6 in podocytes. The authors concluded that there is distinct regulation of the three ER stress response pathways in diabetic nephropathy, and that loss of XBP1 and induction of ATF6 in podocytes are sufficient to activate a maladaptive UPR that is causally linked to diabetic nephropathy24.
Interaction of the UPR with autophagy in diabetes has been addressed in some previous studies12,47,48,49. In one study, the basal level of autophagy in podocytes was reduced in mice with STZ-induced diabetes, as reflected by reductions in LC3-II and other autophagy pathway components50. Treatment of STZ-induced diabetic mice with a chemical chaperone attenuated albuminuria, improved glomerular histopathology and restored autophagy50. The authors proposed that in diabetic nephropathy, podocytes are subjected to ER stress and autophagy is impaired, whereas restoration of autophagy attenuates podocyte injury. Two other studies made similar conclusions51,52. However, deletion of Atg5 autophagy component in podocytes resulted in accelerated podocyte injury and albuminuria in STZ diabetic nephropathy53. Interestingly, a similar phenotype developed after deletion of Atg5 in glomerular endothelial cells. Thus, autophagy appeared to be a key protective mechanism in both cell types of the glomerular capillary wall. Recently, it was shown that type 2 diabetes increased expression of the TRPC6 channel in podocytes in mice and reduced autophagy, while inhibition of calpain (a cysteine protease) normalized autophagy and reduced albuminuria54. Finally, it should be noted that while a majority of studies have focused on the podocyte, a recent study has demonstrated that CD248 (endosialin) is upregulated in mesangial cells in murine types 1 and 2 diabetic nephropathy55. This resulted in impaired glomerular XBP1 splicing by IRE1α and induction of CHOP. Global KO of CD248 attenuated glomerular injury, including albuminuria, supporting the view that communication among glomerular cell types mediates diabetic nephropathy.
Use of chemical chaperones to improve protein folding has provided further evidence for ER stress in diabetic nephropathy. In type 2 diabetic models in db/db mice, chemical chaperones improved blood pressure, fasting plasma glucose levels and insulin tolerance. Chemical chaperones decreased albuminuria, attenuated mesangial expansion and prevented podocyte apoptosis. The effects were paralleled by decreasing levels of ER stress parameters4,51,52. These studies demonstrate that chemical chaperones can modulate diabetic nephropathy; however, the extent to which the positive effects of the drugs are related to improvements in glucose control versus renal ER stress has not been established conclusively.
The epidermal growth factor receptor inhibitor erlotinib slowed the progression of STZ-induced diabetic nephropathy in mice, including reduced albuminuria and histological injury. These effects of erlotinib were associated with reduced expression of ER stress markers, and increased expression of autophagy-associated proteins in glomeruli and tubules56. There is, however, the possibility to consider that epidermal growth factor receptor inhibitors may be acting by enhancing autophagy, independently of receptor inhibition16.
It is important to establish if pathways mediating experimental diabetic nephropathy are relevant to human disease. Using publicly accessible datasets of glomerular gene expression in human kidney biopsies, we found that diabetic nephropathy is associated with activation of multiple pathways involving the ER, Golgi, ER stress, protein folding and autophagy. A substantial number of genes associated with the ER/UPR or autophagy were induced in diabetic nephropathy, compared with healthy controls. A previous study showed that in human diabetic nephropathy kidney biopsies, mRNAs encoding various ER chaperones were elevated13. In another study, nuclear localization of sXBP1 was reduced in kidney biopsies of patients with diabetic nephropathy, whereas ATF6 and CHOP were increased, compared with healthy controls24. These human studies are limited by relatively small numbers of patients, and there is a lack of uniform criteria for performing kidney biopsies in patients with diabetes; nevertheless, the studies do support activation of the UPR and autophagy.
We used GECs in culture with deletion of IRE1α15 to delineate autophagy pathways. Previously, we demonstrated that induction of ER stress in cultured GECs (with tunicamycin) enhances both the UPR and autophagy. Furthermore, tunicamycin increased LC3, Atg5 and Atg7 mRNAs, as well as total LC3 protein, in an IRE1α-dependent manner, consistent with a transcriptional effect of IRE1α-XBP1 on autophagy15. In the present study, we examined autophagy in the context of diabetes. Unlike diabetes in vivo, exposure of GECs to hyperglycemia did not stimulate autophagy. We then treated GECs with C2-ceramide, since ceramides are a group of bioactive sphingolipids that are elevated in podocytes in diabetes35,36,37,38. Ceramides have multiple actions, which may include promoting lipid accumulation, antagonizing mitochondrial function, and inducing ER stress, autophagy and mitophagy36,37,39,57. Induction of autophagy by ceramides may be secondary to the UPR57, or independent of the UPR, e.g. suppression of mTOR activity, activation of AMP-activated protein kinase via starvation, upregulation of beclin-1, or other mechanisms39. In keeping with enhanced autophagy in diabetes in vivo, we found that in cultured GECs, C2-ceramide stimulated an increase in LC3-II and in total LC3 in an IRE1α-dependent manner. However, in contrast to diabetes in vivo, C2-ceramide did not increase GRP94 or MANF in GECs, suggesting that the effect of C2-ceramide on autophagy was independent of the UPR. Earlier, we showed that GECs with deletion or inhibition of IRE1α displayed a defect in autophagosome biogenesis in response to the mTOR inhibitor rapamycin or to glutamine starvation28. By analogy to C2-ceramide, rapamycin and glutamine starvation did not or only minimally stimulated sXBP1 or MANF production. These results support the view that IRE1α is able to mediate autophagy not only via sXBP1 and the UPR, but also independently of sXBP1.
Studies on diabetes in cultured GECs have yielded inconsistent results. In one study, high glucose concentrations promoted autophagy in cultured GECs53, while in another, levels of autophagy markers were reduced in GECs that were exposed to high glucose50. In some studies, cocktails of free fatty acids, angiotensin II and/or cytokines were added to high glucose medium perhaps to better reflect the diabetic milieu37,38. A limitation of studies in cultured GECs is that these cells may not accurately reflect diabetic nephropathy in vivo, and more complex cell culture models, e.g. co-culture of cell lines, may be required to address mechanistic questions58.
In summary, IRE1α is protective to podocytes in mice with diabetic nephropathy. This is associated with activation of the glomerular UPR and autophagy. The results are consistent with gene expression signatures in human diabetic nephropathy and highlight the potential for therapeutically targeting these pathways.
- The Renal Warrior Project. Join Now
- Source: https://www.nature.com/articles/s41598-024-62599-7