The female patient, born to consanguineous parents (first degree cousins) with a family history of Oculocutaneous Albinism in the mother. This was the mother’s first pregnancy and antental ultrasound scans revealed oligohydramnios and Intra-Uterine Growth Retardation (IUGR). The patient was born prematurely at 36 weeks through an emergency cesarean section due to reduced fetal movement and failed induction. The baby was born weighing 2.0 kg with meconium stained liquor and Apgar scores were 6 and 9 at one and five minutes, respectively. The baby required minimal resuscitation and she was managed on continuous positive airway pressure (CPAP) in the first hour of life; however, within a few hours she deteriorated with bilateral pneumothoraces requiring chest drains, intubation, and ventilation. The patient was started on inhaled nitric oxide for hypoxic respiratory failure, and inotropes due to low blood pressure including dopamine, dobutamine, and epinephrine. The patient remained hypotensive with a mean blood pressure of 15–20 mmHg, which required the addition of hydrocortisone followed by vasopressin to improve her blood pressure. Her oxygen saturation measurements were 35%—45% in 100% FiO2. Supportive measures, including sedation, antibiotics, and fluids were administered. The patient didn’t have any urine output and she developed persistent hypoxia and hypotension, necessitating veno-arterial Extra Corporeal Membrane Oxygenation (ECMO) support on the second day of life, which led to an improvement in her oxygen saturation. However, the patients blood pressure remained low despite the ECMO and continuous inotropic support. While on ECMO, renal replacement therapy (CRRT) was initiated, effectively normalzing the creatinine levels, however the CRRT was discontinued due to the development of hypotension, resulting in progressive edema and fluid overload. Subsequently, the decision was made to decannulate and remove the ECMO support due to a substantial right-sided parenchymal hemorrhage and extra-axial hemorrhage observed on head ultrasound. The patient experienced coagulopathy, manifesting as oozing from the skin and chest tubes requiring multiple Fresh Frozen Plasma (FFP), cryoprecipitate, and red cell transfusions due to low hemoglobin, persistent thrombocytopenia and coagulopathy. On the fourth day, a multi-disciplinary team meeting, with the patient’s parents present, concluded to transition the patient from intensive care to comfort care with no further resuscitation. The patient was extubated the following day and passed away a few hours later.
Imaging studies that were done on the baby included: (1) Echocardiography, which showed a structurally normal heart but was associated with severe persistent pulmonary hypertension of newborn (PPHN) and complete right to left shunting across the ductus arteriosus; (2) Abdominal ultrasound, which showed non-specific bilateral echogenic kidneys; (3) Head ultrasound, which showed large left intra-parenchymal and extra-axial acute bleeding associated with mass effect.
The post-mortem examination revealed mildly hypoplastic kidneys, moderate pulmonary hypoplasia, solid and poorly aerated lungs with diffuse alveolar damage, significantly reduced skull vault mineralization and bony development, indicative features of oligohydramnios sequence. Limbs exhibited some flexion changes, and there were characterestics findings of of Potters’ facies, marked edema, and a structurally normal heart. Histopathology showed changes of renal tubular dysgenesis with the renal cortex containing crowded glomeruli separated by small tubules with distal tubular morphology and absence of proximal tubules (Fig. 1). The proximal tubules should be as numerous as the glomeruli and have plump lining cells with abundant cytoplasm. The medulla appeared largely unremarkable. The family history of parental consanguinity and the severity of symptoms prompted enrolling the family in the Mendelian disease program at Sidra Medicine (Fig. 2a) [13]. Genome sequencing was performed on all family members, and following our in-house analysis pipeline [14], the patient was, initially, found to carry six de novo and nine homozygous rare protein-altering variants, including two that were predicted to lead to loss-of-function (LoF) (Additional file 1). These two include a variant in OR1J4 (c.221C > G, p.Ser74*), an olfactory receptor gene not known to be associated with Mendelian disease, and a nonsense previously unreported variant (NM_000685.5; c.415C > T; p.Arg139*) was identified in the Angiotensin II Receptor Type 1 (AGTR1) gene (Table 1). Importantly, LoF variants in this gene have been associated with renal tubular dysgenesis (MIM# 267,430) [8]. Both parents were heterozygous carriers of the variant (Fig. 2b) and in-silico pathogenicity scores predicted it to be highly damaging (CADD of 39 and GERP of 5.8).
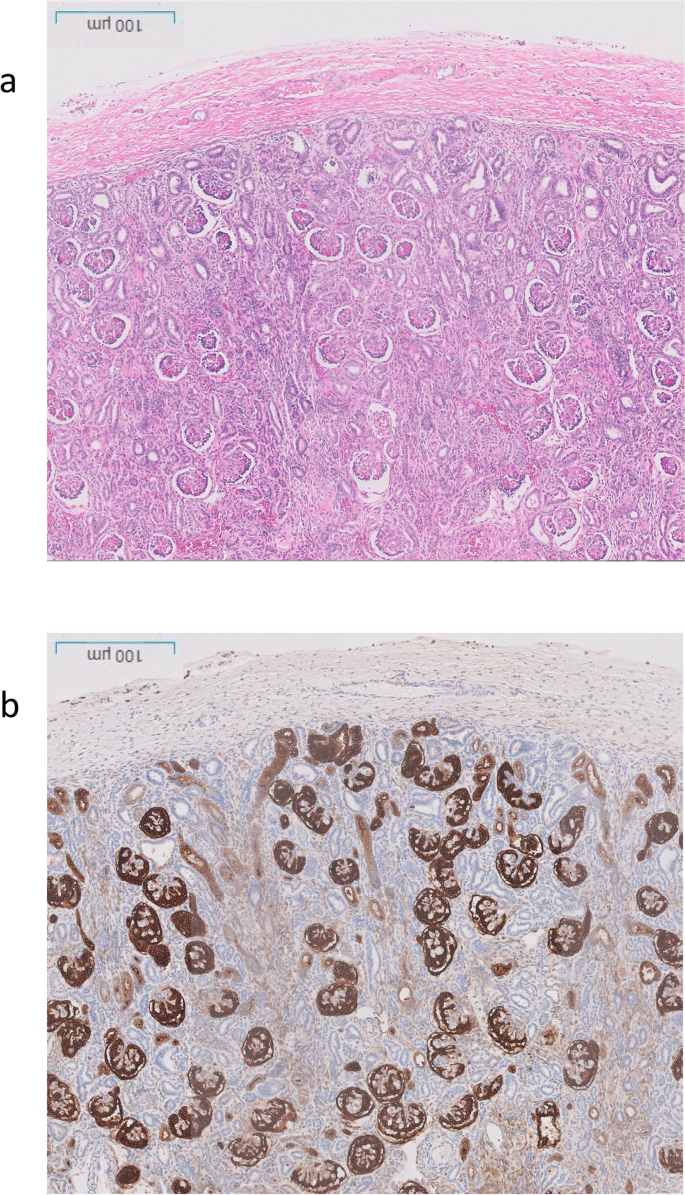
Renal histological characteristics. a H&E renal cortex, showing crowding of the glomeruli, with intervening tubules mainly of distal tubule type, and lack of proximal tubules. b CD10 highlighting the glomeruli and the Bowmans capsule, but normal proximal tubules are not seen, only weak staining of the ureteric buds
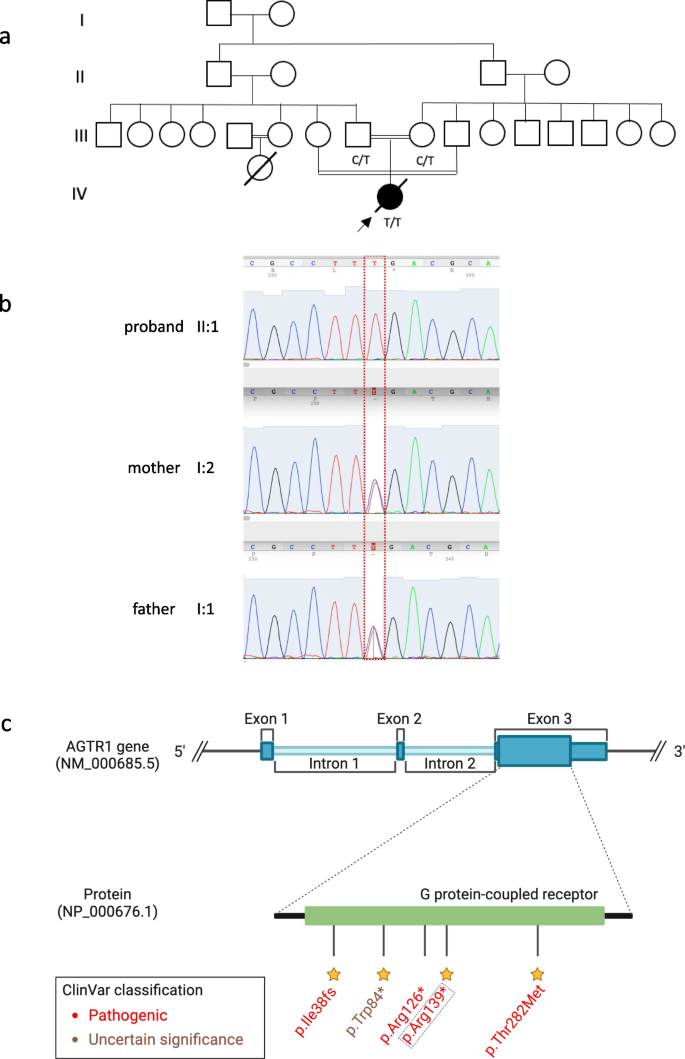
Patient characteristics and genetic findings. a Family pedigree of the patient along with genotypes of the nonesense AGTR1 variant (c.415C > T; p.Arg139*). b Chromatogram of Sanger sequencing showing the variant position and genotypes of the 3 family members. c Schematic of AGTR1 gene body with highlights of protein domains and reported ClinVar variants. The yellow stars refer to the staring system of ClinVar which indicate the review status of the variant
- The Renal Warrior Project. Join Now
- Source: https://bmcnephrol.biomedcentral.com/articles/10.1186/s12882-024-03569-z